Старение характеризуется постепенной потерей организмом физиологической целостности, ведущей к нарушениям его функций и увеличению риска смерти. Это ухудшение является главным фактором риска основных патологий человека, включая рак, диабет, сердечно-сосудистые и нейродегенеративные заболевания. В недалеком прошлом ученым удалось добиться беспрецедентных успехов в изучении процессов старения, особенно с установлением того факта, что скорость старения в некоторой степени контролируется сохранившимися в процессе эволюции генетическими и биохимическими процессами. В данном обзоре перечислены 9 потенциальных ключевых признаков, которые являются общими для процесса старения у различных организмов, с акцентом на млекопитающих. К этим признакам можно отнести: нестабильность генома, укорочение теломер, эпигенетические альтерации, нарушение протеостаза, нарушение распознавания питательных веществ, митохондриальную дисфункцию, клеточное старение, истощение пула стволовых клеток и изменение межклеточного взаимодействия. Основной задачей является определение взаимосвязи между потенциальными ключевыми признаками и их относительным вкладом в старение с конечной целью установить фармацевтические мишени, которые с минимальными побочными эффектами позволят улучшить здоровье человека при изменениях, вызванных старением.

Введение
Старение, в общих чертах определяемое как зависимое от времени снижение функций, затрагивает большинство живых организмов. На протяжении всей истории человечества оно вызывает любопытство и будоражит воображение, хотя и прошло всего 30 лет с начала новой эры в исследовании старения, ознаменованной получением первых долгоживущих линий Caenorhabditis elegans (C. elegans) (Klass, 1983). В наши дни старение является предметом тщательных научных исследований, основанных на расширяющихся знаниях о молекулярных и клеточных основах жизни и заболеваний. В нынешних условиях можно провести множество параллелей между исследованиями старения и онкологическими исследованиями, выполненными в предыдущие десятилетия. Ключевым моментом в области изучения рака стала публикация знаменательной работы, в которой было перечислено шесть важнейших признаков рака (Hanahan and Weinberg, 2000), список которых недавно был увеличен до десяти (Hanahan and Weinberg, 2011). Эта систематизация помогла осмыслить природу рака и механизмы, лежащие в его основе.
На первый взгляд, старение и рак могут показаться противоположными процессами: рак является последствием нарушения механизмов адаптации клетки (в том числе и их чрезмерной активацией), тогда как старение характеризуется их утратой. Но если мы копнем глубже, то увидим, что старение и рак имеют общие корни. Общепризнанно, что накопление клеточных повреждений с течением времени является основной причиной старения (Gems and Partridge, 2013; Kirkwood, 2005; Vijg and Campisi, 2008).
В то же время клеточные повреждения могут приводить к появлению аномальных преимуществ у клеток, которые в итоге могут стать раковыми. Таким образом, рак и старение могут рассматриваться как два разных проявления одного и того же процесса – накопления клеточных повреждений. Вдобавок некоторые ассоциированные со старением патологии, такие как атеросклероз и воспаление, подразумевают неконтролируемое клеточное деление и гиперфункцию (Blagosklonny, 2008). На основании этой концепции в области исследования старения возникли некоторые вопросы, касающиеся физиологического источника повреждений, вызывающих старение, компенсаторных реакций, направленных на восстановление гомеостаза, взаимосвязи между различными типами повреждений и компенсаторными реакциями, а также возможности экзогенных вмешательств, нацеленных на замедление старения.
В данной работе мы предприняли попытку определить и классифицировать основные клеточные и молекулярные признаки старения. Мы предлагаем девять основных признаков старения, которые, как считается, вносят вклад в этот процесс и определяют его фенотип (Рисунок 1). Учитывая сложность вопроса, мы сконцентрируемся на современном понимании старения млекопитающих, принимая во внимание результаты передовых исследований более простых организмов (Gems and Partridge, 2013; Kenyon, 2010). Каждый ключевой признак должен удовлетворять следующим критериям: (1) он должен наблюдаться при нормальном старении; (2) его экспериментальное усиление должно приводить к ускоренному старению; (3) его экспериментальное ослабление должно замедлять развитие нормального старения и тем самым увеличивать здоровую продолжительность жизни. Все предложенные признаки удовлетворяют этим критериям в разной степени, что будет обсуждаться для каждого признака в отдельности. Последний критерий является самым труднодостижимым, даже если свести его к какому-то одному аспекту старения. По этой причине мероприятия, которые способны замедлять старение, влияют не на все ключевые признаки. Это противоречие разрешается избыточным количеством связей между ключевыми признаками старения, таким образом, экспериментальное улучшение одного может повлиять на остальные.
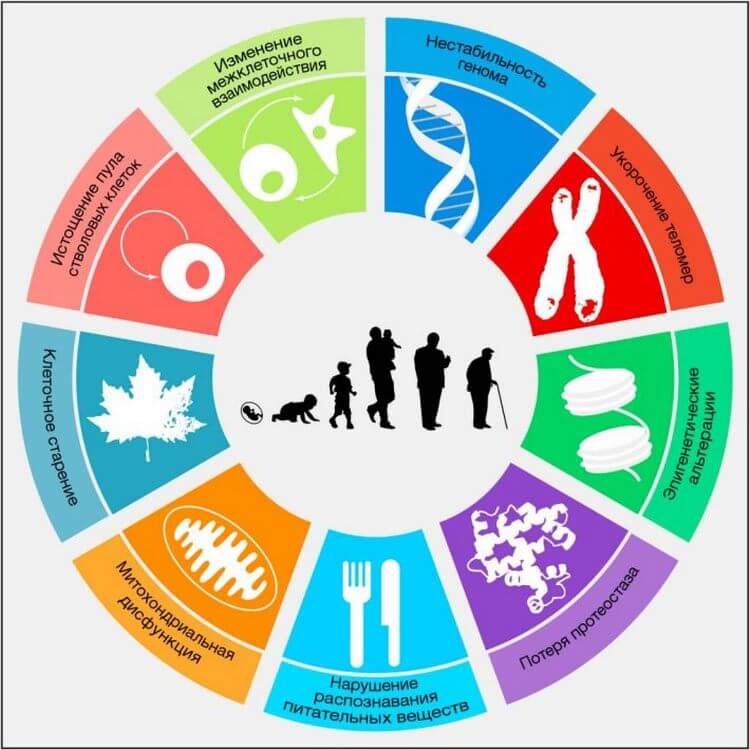
Рисунок 1. Ключевые признаки старения
На схеме изображены девять характерных признаков, описанных в данном обзоре: нестабильность генома, укорочение теломер, эпигенетические альтерации, нарушение протеостаза, нарушение распознавания питательных веществ, митохондриальная дисфункция, клеточное старение, истощение пула стволовых клеток, а также изменение межклеточного взаимодействия.
Нестабильность генома
Одним из общих звеньев процесса старения является накопление генетических повреждений на протяжении всей жизни (Moskalev et al., 2012) (Рисунок 2А). Более того, многие болезни преждевременного старения, в частности синдром Вернера и синдром Блума, являются следствием повышенного накопления повреждений ДНК (Burtner and Kennedy, 2010), хотя уместность этих и других прогероидных синдромов в отношении нормального старения остается неясной, отчасти потому что они повторяют только некоторые черты старения. Целостность и стабильность ДНК постоянно подвергается опасности как со стороны экзогенных (физических, химических и биологических агентов), так и со стороны эндогенных факторов, таких как ошибки репликации ДНК, реакции спонтанного гидролиза и активные формы кислорода (АФК) (Hoeijmakers, 2009). Генетические нарушения, возникающие из-за внутренних и внешних повреждений, довольно разнообразны и включают точечные мутации, транслокации, укорочение и удлинение хромосом, укорочение теломер и повреждение генов, вызванное интеграцией вирусов или транспозонов. Чтобы свести к минимуму эти повреждения, организмы выработали сложную сеть механизмов репарации ДНК, которые в совокупности способны справиться с большинством повреждений, нанесенных ядерной ДНК (Lord and Ashworth, 2012). К системам поддержания стабильности генома относятся специальные механизмы для стабилизации необходимой длины и функциональности теломер (это является частью другого ключевого признака, указанного ниже) и для сохранения целостности митохондриальной ДНК (мтДНК) (Blackburn et al., 2006; Kazak et al., 2012). Помимо прямых повреждений ДНК, к дестабилизации генома и преждевременному старению могут приводить также дефекты ядерной архитектуры, известные как ламинопатии (Worman, 2012).
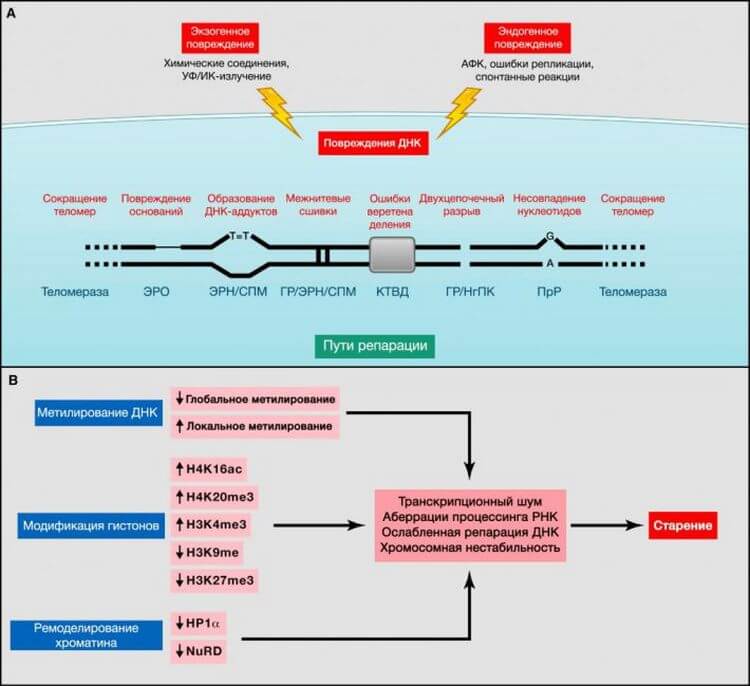
Рисунок 2. Геномные и эпигенетические альтерации
(A) Геномная нестабильность и укорочение теломер. Эндогенные или экзогенные агенты могут стимулировать множество повреждений ДНК, схематично представленных на одной хромосоме. Такие повреждения могут быть устранены различными механизмами. Чрезмерное повреждение или же недостаточная репарация ДНК способствуют старению. Обратите внимание на то, что и ядерная, и митохондриальная (не представленная здесь) ДНК подвергаются возрастным геномным альтерациям. ЭРО, эксцизионная репарация оснований; ГР, гомологичная рекомбинация; ЭРН, эксцизионная репарация нуклеотидов; НгПК, негомологичное присоединение концов; ПрР, пострепликативная репарация; АФК, активные формы кислорода; СПМ, синтез на повреждённой матрице; КТВД, контрольная точка веретена деления (Vijg, 2007).(B) Эпигенетические альтерации. Изменения метилирования ДНК или ацетилирования и метилирования гистонов, а также других ассоциированных с хроматином белков, могут вызвать эпигенетические альтерации, способствующие процессу старения.
Ядерная ДНК
С возрастом в клетках человека и модельных организмов накапливаются соматические мутации (Moskalev et al., 2012). Со старением ассоциированы и другие формы повреждений ДНК: анеуплоидии и вариации числа копий генов (Faggioli et al., 2012; Forsberg et al., 2012). Также задокументирован высокий клональный мозаицизм больших хромосомных аномалий (Jacobs et al., 2012; Laurie et al., 2012). Все эти формы перестроек в ДНК могут влиять на основные гены и транскрипционные пути, что приводит к появлению не справляющихся со своими функциями клеток, которые могут подвергать опасности тканевой и организменный гомеостаз, если не удаляются апоптозом или не приобретают сенесцентный фенотип. Это особенно важно в отношении того, как повреждение ДНК влияет на функциональные возможности стволовых клеток, мешая им играть свою роль в обновлении тканей (Jones and Rando, 2011; Rossi et al., 2008) (См. «истощение стволовых клеток»).
Свидетельства причинно-следственной связи между увеличением количества геномных повреждений на протяжении жизни и старением вытекают из исследований, проведенных на мышах и людях, которые показывают, что неполноценность механизмов репарации ДНК ускоряет старение у мышей и лежит в основе человеческих прогероидных синдромов, таких как синдром Вернера, синдром Блума, пигментная ксеродерма, трихотиодистрофия, синдром Коккейна и синдром Секкеля (Gregg et al., 2012; Hoeijmakers, 2009; Murga et al., 2009). Более того, у трансгенных мышей с чрезмерной экспрессией BubR1 — компонента контрольной точки митотического цикла, контролирующего точную сегрегацию хромосом — отмечается высокая устойчивость к анеуплоидии и опухолевой трансформации, а также увеличение продолжительности здоровой жизни (Baker et al., 2013). Последние результаты служат экспериментальным доказательством того, что искусственное укрепление механизмов репарации ядерной ДНК способно замедлять старение.
Митохондриальная ДНК
Возрастные мутации и делеции мтДНК также вносят вклад в старение (Park and Larsson, 2011). мтДНК считается основной мишенью возрастных соматических мутаций, которые развиваются из-за действия окислительной микросреды митохондрий, отсутствия защитных гистонов мтДНК и малой эффективности механизмов репарации в сравнении с ядерной ДНК (Linnane et al., 1989). Вовлеченность мутаций мтДНК в процесс старения подвергалась сомнению из-за большого количества копий митохондриального генома, которые допускают сосуществование мутантного генома и генома «дикого типа» в одной и той же клетке (этот феномен иначе именуется «гетероплазмией»). Однако анализы одиночных клеток показали, что, несмотря на низкий общий уровень мутаций мтДНК, мутационный груз отдельных стареющих клеток становится значительным и может достигать состояния гомоплазмии, при котором превалирует мутантный геном (Khrapko et al., 1999). Интересно то, что, вопреки имеющимся ожиданиям, большинство мутаций мтДНК в зрелых или сенесцентных клетках вызваны ошибками репликации в начале жизни, а не окислительными повреждениями. Эти мутации могут претерпевать поликлональную экспансию и вызывать нарушение функции дыхательной цепи в разных тканях (Ameur et al., 2011). Исследования пациентов с ускоренным старением и ВИЧ-инфицированных пациентов, получающих антиретровирусные препараты, которые препятствуют репликации мтДНК, свидетельствуют в пользу концепции клональной экспансии появившихся на ранних этапах жизни мутаций мтДНК (Payne et al., 2011).
Первое свидетельство того, что повреждение мтДНК играет важную роль в старении человека и развитии возрастных заболеваний, было получено в ходе изучения мультисистемных заболеваний человека, вызванных мутациями мтДНК, которые частично являются фенокопиями старения (Wallace, 2005). Следующее свидетельство было получено в ходе исследования мышей с недостаточностью митохондриальной γ-ДНК-полимеразы. Эти мутантные мыши демонстрируют признаки ускоренного старения и имеют более короткую продолжительность жизни в ассоциации со случайными точечными мутациями и делециями мтДНК (Kujoth et al., 2005; Trifunovic et al., 2004; Vermulst et al., 2008). Функции митохондрий клеток этих мышей нарушены, однако нарушения не сопровождаются увеличением продукции АФК (Edgar et al., 2009; Hiona et al., 2010). Более того, стволовые клетки данных прогероидных мышей особенно чувствительны к накоплению мутаций в мтДНК (Ahlqvist et al., 2012) (См. «Истощение стволовых клеток»). Необходимы дальнейшие исследования, которые позволят определить, способны ли генетические манипуляции, снижающие количество мутаций мтДНК, увеличивать продолжительность жизни.
Ядерная архитектура
Дефекты ядерной ламины могут вызывать нестабильность генома (Dechat et al., 2008). Ядерные белки промежуточных филаментов (ламины) составляют основную часть ядерной ламины и участвуют в поддержании сохранности генетического материала, представляя собой каркас для крепления хроматина и белковых комплексов, которые регулируют стабильность генома (Gonzalez-Suarez et al., 2009; Liu et al., 2005). Ядерная ламина привлекла внимание исследователей в области старения после открытия мутаций в генах, кодирующих белковые компоненты этих структур или факторов, влияющих на их созревание и динамику и вызывающих такие прогероидные синдромы, как синдром Хатчинсона-Гилфорда и синдром Нестора-Гильермо (СПХГ и СПНГ соответственно) (Cabanillas et al., 2011; De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). Изменения ядерной ламины и продукция аберрантной изоформы преламина А (иначе называемой — «прогерин») выявляются и при нормальном старении человека (Ragnauth et al., 2010; Scaffidi and Misteli, 2006). Дисфункция теломер также увеличивает продукцию прогерина в нормальных человеческих фибробластах в культуре in vitro, что дает основания предположить наличие дополнительных связей между поддержанием длины теломер и экспрессией прогерина при нормальном старении (Cao et al., 2011). В дополнение к этим возрастным изменениям в ламинах типа А, уровень ламинов типа В1 при старении клеток уменьшается, что указывает на их полезность в качестве биомаркера этого процесса (Freund et al., 2012; Shimi et al., 2011).
Животные и клеточные модели облегчили идентификацию стрессовых метаболических путей, развивающихся под действием нарушения структуры ядерной ламины при СПХГ. Эти пути сочетают активацию p53 (Varela et al., 2005), дерегуляцию соматотропной оси (Marin˜ o et al., 2010) и истощение стволовых клеток взрослого организма (Espada et al., 2008; Scaffidi and Misteli, 2008). Основанием полагать, что нарушения в ядерной ламине вносят вклад в ускоренное старение, служит тот факт, что снижение уровня преламина А или прогерина отсрочивает начало проявления прогероидных симптомов и увеличивает продолжительность жизни в мышиных моделях СПХГ. Этого можно достичь системным вводом антисмысловых олигонуклеотидов, ингибиторов фарнезилтрансферазы или комбинацией статинов и аминобисфосфонатов (Osorio et al., 2011; Varela et al., 2008; Yang et al., 2006). Восстановление соматотропной оси при помощи гормональной терапии или ингибирования NF-kB сигналинга также увеличивает продолжительность жизни прогероидных мышей (Marin˜ o et al., 2010; Osorio et al., 2012). Кроме того, была разработана стратегия, основанная на гомологичной рекомбинации и направленная на устранение мутаций гена LMNA в индуцированных плюрипотентных стволовых клетках (иПСК), полученных от пациентов с СПХГ. Этот принцип открывает широкие перспективы клеточной терапии в будущем (Liu et al., 2011b). Необходимы дальнейшие исследования, которые позволят подтвердить, что укрепление ядерной архитектуры способно отсрочить нормальное старение.
В общих чертах
Существует множество доказательств того, что старение сопровождается повреждениями генома, а его искусственные повреждения способны ускорить старение. Отмечено, что усиление работы механизмов, обеспечивающих надлежащую сегрегацию хромосом, увеличивает продолжительность жизни млекопитающих (Baker et al., 2013). Кроме того, в частном случае прогерии, ассоциированной с дефектами ядерной архитектуры, существуют доказанные терапевтические методики, способные отсрочить преждевременное старение. Необходимо изучить похожие средства, чтобы найти способы воздействия на другие аспекты стабильности ядерного и митохондриального генома, например такие, как репарация ДНК, которая может оказывать положительное влияние на нормальное старение (теломеры представляют особый случай и обсуждаются отдельно).
Укорочение теломер
Накопление повреждений ДНК с возрастом беспорядочно воздействует на геном, однако такие участки хромосом, как теломеры, с возрастом особенно подвержены изнашиванию (Blackburn et al., 2006) (Рисунок 2А). У репликативных ДНК-полимераз отсутствует способность полностью реплицировать терминальные концы линейных молекул ДНК, — функция, свойственная специализированной ДНК-полимеразе, также известной как теломераза. Однако большинство соматических клеток млекопитающих не экспрессируют теломеразу, что ведет к постепенно нарастающей потере последовательностей на концах хромосом, защищающих теломеры. Укорочение теломер объясняет ограниченную пролиферативную способность некоторых типов культур клеток, выращиваемых in vitro, также известную как феномен репликативного старения или предел Хейфлика (Hayflick and Moorhead, 1961; Olovnikov, 1996). Действительно, эктопической экспрессии теломеразы будет достаточно, чтобы даровать бессмертие смертным клеткам, не вызывая опухолевой трансформации (Bodnar et al., 1998). Важно то, что укорочение теломер при нормальном старении наблюдается как у людей, так и у мышей (Blasco, 2007).
Теломеры связаны с характерным мультипротеиновым комплексом, известным как шелтерин (Shelterin) (Palm and de Lange, 2008). Основная функция комплекса — это защита теломер от ферментов репарации ДНК. Иначе теломеры будут «починены» как разрывы ДНК, что приведет к слиянию хромосом. Из-за ограниченной репарации ДНК, повреждения в больших количествах накапливаются в теломерах и индуцируют старение и/или апоптоз (Fumagalli et al., 2012; Hewitt et al., 2012).
Недостаточность теломеразы у людей ассоциирована с ранним развитием таких заболеваний, как фиброз легких, врожденный дискератоз и апластическая анемия, которые включают потерю регенеративных способностей различных тканей (Armanios and Blackburn, 2012). Потеря теломерами защитных «колпачков» и бесконтрольное слияние хромосом также могут возникнуть при неправильной работе компонентов шелтерина (Palm and de Lange, 2008). Мутации шелтерина были обнаружены в некоторых случаях апластической анемии и врожденного дискератоза (Savage et al., 2008; Walne et al., 2008; Zhong et al., 2011). При моделировании случаев потери компонентами шелтерина своих функций отмечается резкое снижение регенеративной способности тканей и ускоренное их старение. Этот феномен может наблюдаться и при нормальной длине теломер (Martı´nez and Blasco, 2010).
Генетически модифицированные модельные животные помогли установить связь между потерей теломер, клеточным старением и старением организма. Так, мыши с укороченными теломерами живут меньше, а с более длинными – дольше (Armanios et al., 2009; Blasco et al., 1997; Herrera et al., 1999; Rudolph et al., 1999; Toma´ s-Loba et al., 2008). Последние открытия также указывают на то, что старение может быть обращено вспять активацией теломеразы. В частности, раннее старение теломеразо-дефицитных мышей можно предотвратить генетической реактивацией их теломеразы (Jaskelioff et al., 2011). Кроме того, у взрослых мышей дикого типа можно замедлить нормальное физиологическое старение путем системной вирусной трансдукции теломеразы, что не увеличит вероятность развития рака (Bernardes de Jesus et al., 2012). Недавний мета-анализ подтвердил существование корреляции между смертностью и короткой длиной теломер у людей, особенно в молодом возрасте (Boonekamp et al., 2013).
В общих чертах
Нормальное старение у млекопитающих сопровождается укорочением теломер. Более того, патологическая дисфункция теломер ускоряет старение у мышей и человека, тогда как экспериментальная стимуляция теломеразы способна замедлить старение мышей. Таким образом, данный признак соответствует всем критериям ключевого признака старения.
Эпигенетические альтерации
В течение жизни во всех клетках и тканях происходят разнообразные эпигенетические альтерации (Talens et al., 2012) (Рисунок 2B). Эти модификации включают изменение паттерна метилирования ДНК, посттрансляционную модификацию гистонов и ремоделирование хроматина. Повышенное ацетилирование гистона H4K16, триметилирование H4K20 или H3K4, а также снижение метилирования H3K9 или H3K27 являются ассоциированными с возрастом признаками (Fraga and Esteller, 2007; Han and Brunet, 2012). К многочисленным ферментным системам, отвечающим за генерацию и поддержание эпигенетических паттернов, относятся ДНК метилтрансферазы, гистоновые ацетилазы, деацетилазы, метилазы, деметилазы, а также белковые комплексы, вовлеченные в ремоделирование хроматина.
Модификации гистонов
Метилирование гистонов соответствует критериям ключевого признака старения у беспозвоночных. Делеция компонентов комплексов метилирования гистонов (для H3K4 и для H3K27) увеличивает продолжительность жизни нематод и мух, соответственно (Greer et al., 2010; Siebold et al., 2010). Кроме того, ингибирование гистоновых деметилаз (для H3K27) может увеличивать продолжительность жизни у червей за счет влияния на компоненты ключевых путей долголетия, таких как сигнальный путь инсулин/ИФР-1 (Jin et al., 2011). До конца не ясно, могут ли манипуляции с гистон-модифицирующими ферментами влиять на старение, используя исключительно эпигенетические механизмы, накладывающиеся на репарацию ДНК и стабильность генома, или же они воздействуют посредством транскрипционных изменений, затрагивающих метаболические и сигнальные пути вне ядра.
В качестве потенциальных факторов, замедляющих старение, рассматриваются НАД-зависимые протеин-деацетилазы и АДФ-рибозил трансферазы из семейства сиртуинов. Интерес к данному семейству белков начался с серии работ на дрожжах, мухах и червях, в которых было отмечено, что единственный ген сиртуина в этих организмах, называемый Sir2, имеет примечательную активность в отношении долголетия (Guarente, 2011). Увеличение репликативной продолжительности жизни при сверхэкспрессии Sir2 было впервые зарегистрировано у Saccharomyces cerevisiae (Kaeberlein et al., 1999), позже подобный эффект наблюдался у модельных беспозвоночных организмов при чрезмерной экспрессии ортологов у червей (sir-2.1) и мух (dSir2) (Rogina и Helfand, 2004; Tissenbaum и Guarente, 2001). Однако недавно эти открытия вызвали ряд вопросов в связи с докладом, показавшим, что наблюдаемое увеличение продолжительности жизни в работе на червях и мухах было обусловлено существенными различиями в генетическом контексте, а не сверхэкспрессией sir-2.1 или dSir2 соответственно (Burnett et al., 2011). В самом деле, тщательная переоценка показала, что сверхэкспрессия sir-2.1 приводит к умеренному увеличению продолжительности жизни только у C. elegans (Viswanathan and Guarente, 2011).
Некоторые из 7 паралогов сиртуина, присущих млекопитающим, могут влиять на различные аспекты старения у мышей (Houtkooper et al., 2012; Sebastia´ n et al., 2012). В частности трансгенная сверхэкспрессия SIRT1, который является ближайшим гомологом Sir2 беспозвоночных у млекопитающих, улучшает общее состояние в процессе старения, но не влияет на долголетие (Herranz et al., 2010). Механизмы, которые обеспечивают положительные эффекты SIRT1, сложны и взаимосвязаны, и включают повышение стабильности генома (Oberdoerffer et al., 2008; Wang et al., 2008), а также усиление эффективности метаболизма (Nogueiras et al., 2012) (см. ‘‘Нарушение распознавания питательных веществ’’). Более убедительные доказательства того, что сиртуины играют положительную роль в долголетии, были получены для SIRT6, который регулирует стабильность генома, NF-kB сигналинг и гомеостаз глюкозы через деацетилирование H3K9 (Kanfi et al., 2010; Kawahara et al., 2009; Zhong et al., 2010). Дефицитные по SIRT6 мыши стареют быстрее контрольных животных (Mostoslavsky et al., 2006), тогда как самцы трансгенных мышей со сверхэкспрессией SIRT6 имеют большую, чем контрольные животные продолжительность жизни, ассоциированную со снижением концентрации ИФР-1 и других компонентов сигнального каскада ИФР-1 в сыворотке крови (Kanfi et al., 2012). Интересно, что располагающийся в митохондриях сиртуин SIRT3, по результатам исследований, был ответственен за некоторые из положительных эффектов ограничения калорийности питания, способствовавших долголетию, хотя эти эффекты развивались не вследствие гистоновых модификаций, а скорее вследствие деацетилирования митохондриальных белков (Someya et al., 2010). Совсем недавно было показано, что сверхэкспрессия SIRT3 улучшает регенеративные способности стареющих гемопоэтических стволовых клеток (Brown et al., 2013). Таким образом, у млекопитающих как минимум три члена семейства сиртуинов (SIRT1, SIRT3 и SIRT6) вносят вклад в здоровое старение.
Метилирование ДНК
Связь между метилированием ДНК и старением неоднозначна. Ранние работы выявили ассоциированное с возрастом системное гипометилирование, однако последующий анализ показал, что несколько локусов, в том числе и локусы различных генов опухолевой супрессии и генов-мишеней Polycomb, с возрастом, наоборот, подвергаются избыточному метилированию (Maegawa et al., 2010). Клетки пациентов и мышей с синдромами прогерии имеют такие же паттерны метилирования ДНК и модификаций гистонов, как и у нормально стареющих клеток (Osorio et al. 2010; Shumaker et al. 2006). Все эти эпигенетические дефекты или эпимутации, накапливаясь с возрастом, специфически воздействуют на поведение и функции стволовых клеток (Pollina and Brunet, 2011) (см. “Истощение стволовых клеток”). Тем не менее, не было проведено прямого эксперимента, показывающего, что продолжительность жизни организма может быть увеличена при изменении паттернов метилирования ДНК.
Ремоделирование хроматина
ДНК- и гистон-модифицирующие ферменты действуют вместе с ключевыми хромосомными белками, такими как гетерохроматиновый белок 1a (HP1a), и факторами ремоделирования хроматина, такими как белки группы Polycomb или комплекс NuRD, уровни которых снижаются при нормальном и патологическом старении клеток (Pegoraro et al. 2009; Pollina and Brunet, 2011). В дополнение к описанным выше эпигенетическим модификациям в гистонах и метилированию ДНК, альтерации в данных эпигенетических факторах определяют такие изменения в архитектуре хроматина, как глобальная потеря и перераспределение гетерохроматина, которые являются характерными признаками старения (Oberdoerffer and Sinclair, 2007; Tsurumi and Li, 2012). Связь этих изменений хроматина и старения подтверждается открытием того, что мутантные мухи с потерей функции в HP1a имеют более короткую продолжительность жизни, тогда как сверхэкспрессия этого гетерохроматинового белка увеличивает продолжительность жизни у мух и задерживает ухудшение работы мышц, характерное для старения (Larson et al. 2012).
Зависимость между формированием повторов ДНК и хромосомной стабильностью подтверждает функциональную значимость эпигенетических изменений хроматина при старении. В частности, сборка гетерохроматина в перицентрических регионах требует триметилирования гистонов H3K9 и H4K20, а также связывания HP1a, и поддерживает хромосомную стабильность (Schotta et al. 2004). Теломерные повторы млекопитающих обогащены этими модификациями хроматина, что указывает на то, что концы хромосом собираются в гетерохроматиновые домены (Blasco, 2007b; Gonzalo et al., 2006). Субтеломерные регионы также имеют черты конституитивного гетерохроматина, включающие триметилирование гистонов H3K9 и H4K20, связывание HP1a и гиперметилирование ДНК. Таким образом, эпигенетические альтерации могут напрямую вмешиваться в регуляцию длины теломер — одного из ключевых признаков старения.
Транскрипционные изменения
Усиление транскрипционного шума (Bahar et al., 2006), аберрантный синтез и созревание мРНК (Harries et al., 2011; Nicholas et al., 2010) − неизбежные спутники старения. Сравнения молодых и старых тканей нескольких видов с использованием микрочипов помогли обнаружить накапливающиеся с возрастом транскрипционные изменения в генах, кодирующих ключевые компоненты воспалительного процесса, а также деградации митохондрий и лизосом (de Magalhaes et al., 2009). Эти возрастные изменения профиля транскрипции также влияют на некодирующие РНК, включающие класс миРНК (геро-миРНК), который ассоциирован со старением и влияет на продолжительность жизни, воздействуя на компоненты сигнальных путей, регулирующих продолжительность жизни или поведение стволовых клеток (Boulias and Horvitz, 2012; Toledano et al., 2012; Ugalde et al., 2011). Исследования мутаций с приобретением или потерей функции подтвердили способность некоторых миРНК влиять на продолжительность жизни Drosophila melanogaster и C. elegans (Liu et al., 2012; Shen et al., 2012; Smith-Vikos and Slack, 2012).
Обратимость эпигенетических изменений
В отличие от мутаций ДНК, эпигенетические изменения являются, по крайней мере теоретически, обратимыми, что делает возможным создание новых средств против старения (Freije and Lopez-Otin, 2012; Rando and Chang, 2012). Восстановление физиологического ацетилирования гистона Н4 с использованием ингибиторов гистоновых деацетилаз позволяет избежать проявления возраст-зависимых нарушений памяти у мышей. Это указывает на то, что восстановление эпигенетических нарушений может иметь нейропротекторный эффект. Ингибиторы гистоновых ацетилтрансфераз также уменьшают внешние проявления старения прогероидных мышей и увеличивают продолжительность их жизни (Krishnan et al., 2011). Более того, недавнее открытие эпигенетического наследования долголетия из поколения в поколение у C. elegans дает основание полагать, что манипуляция со специфическими модификациями хроматина у родителей может индуцировать эпигенетическую память о долголетии у их потомков (Greer et al., 2011). Активаторы гистоновых деацетилаз, предположительно, могут увеличивать продолжительность жизни принципиально схожим с ингибиторами гистоновых ацетилтрансфераз образом. Ресвератрол детально изучается в отношении старения, и среди многочисленных механизмов его действия имеется повышение активности SIRT1 и другие эффекты, ассоциированные с энергетическим дефицитом (см. «Митохондриальная дисфункция).
В общих чертах
Существуют многочисленные свидетельства, дающие основание полагать, что старение сопровождается эпигенетическими изменениями, которые могут провоцировать прогероидные синдромы в модельных организмах. SIRT6 служит примером эпигенетически значимого фермента, и потеря им активности в результате мутации сокращает продолжительность жизни мыши, а усиление активности – увеличивает (Kanfi et al., 2012; Mostoslavsky et al., 2006). Все вместе эти работы свидетельствуют, что понимание и обработка эпигенома являются многообещающими в отношении борьбы с возраст-зависимыми патологиями и увеличения здоровой продолжительности жизни.
Нарушение протеостаза
Старение и некоторые возраст-зависимые болезни связаны с нарушением белкового гомеостаза или протеостаза (Powers et al., 2009) (Рисунок 3). Все клетки с успехом используют набор механизмов контроля качества, направленных на сохранение стабильности и функциональности их протеомов. Протеостаз включает механизмы стабилизации правильно уложенных белков, из них наиболее известный – система белков теплового шока, а также механизмы деградации белков протеосомами или лизосомами (Hartl et al., 2011; Koga et al., 2011; Mizushima et al., 2008). Кроме того, существуют регуляторы возраст-зависимой протеотоксичности, такие как MOAG-4, которые действуют альтернативными путями, отличными от молекулярных шаперонов и протеаз (van Ham et al., 2010). Все эти системы действуют слаженно, чтобы восстановить структуру неправильно сложенного полипептида или разрушить его, предотвращая накопление поврежденных компонентов и обеспечивая постоянное обновление внутриклеточных белков. Многие работы показали, что протеостаз нарушается при старении (Koga et al., 2011). Кроме того, постоянная экспрессия несложенных, сложенных неправильно или агрегированных белков вносит вклад в развитие некоторых возрастных патологий, таких как болезнь Альцгеймера, болезнь Паркинсона и катаракта (Powers et al., 2009).
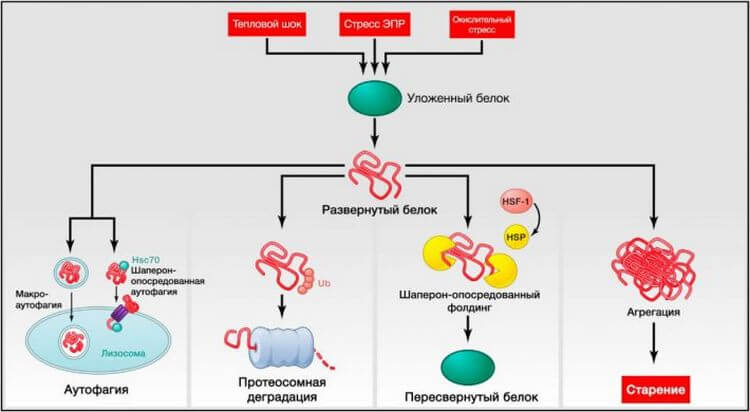
Рисунок 3. Нарушение протеостаза
Эндогенный и экзогенный стресс вызывает разворачивание белков (или нарушает их правильную укладку во время синтеза). Несвернутые белки обычно претерпевают рефолдинг белками теплового шока (БТШ) или служат мишенью деструкции убиквитин-протеосомных или лизосомальных (аутофагических) путей. Аутофагические пути включают узнавание несвернутых белков шапероном Hsc70 и их последующий перенос в лизосомы (шаперон-опосредованная аутофагия) или разрушение поврежденных белков и органелл в аутофагосомах, позже соединяющихся с лизосомами (макроаутофагия). Неспособность рефолдинга или деградации несвернутых белков может привести к их накоплению и агрегации, приводящей к протеотоксическим эффектам.
Шаперон-опосредованный фолдинг и стабильность белка
При старении в значительной степени нарушен вызываемый стрессом синтез цитозольных и органелло-специфичных шаперонов (Calderwood et al., 2009). Многочисленные исследования на модельных организмах подтверждают влияние ухудшения работы шаперонов на продолжительность жизни. В частности, долгоживущими являются трансгенные черви и мухи со сверхэкспрессией шаперонов (Morrow et al., 2004; Wlaker and Lithgow., 2003). Мутантные мыши с дефицитом кошаперона из семейства белков теплового шока стареют быстрее, тогда как линии долгоживущих мышей демонстрируют значительное повышение экспрессии некоторых белков теплового шока (Min et al., 2008; Swindell et al., 2009). Более того, активация основного регулятора ответа на тепловой шок – транскрипционного фактора HSF-1 – увеличивает продолжительность жизни и термотолерантность у нематод (Chiang et al., 2012; Hsu et al., 2003). В то же время компоненты, связывающие амилоид, также могут поддерживать протеостаз при старении и увеличивать продолжительность жизни (Alavez et al., 2011). В клетках млекопитающих деацетилирование HSF-1 белком SIRT1 обуславливает трансактивацию таких генов теплового шока, как Hsp70, тогда как ингибирование SIRT1 ослабляет ответ на тепловой шок (Westerheide et al., 2009).
Протеолитические системы
При старении снижается активность двух основных протеолитических систем, вовлеченных в контроль качества белков – лизосомальной системы, осуществляющей аутофагию, и убиквитин-протеасомной системы (Rubinsztein et al., 2011; Tomaru et al., 2012), что подтверждает особую роль протеостаза в старении.В отношении аутофагии, трансгенные мыши с дополнительной копией опосредуемого шаперонами рецептора аутофагии LAMP2a с возрастом сохраняют функции печени на хорошем уровне и не сталкиваются с возраст-зависимым снижением активности аутофагии (Zhang и Cuervo, 2008). Внедрение химических индукторов макроаутофагии (другой тип аутофагии, отличный от опосредуемой шаперонами аутофагии) вызвало необычайный интерес после открытия того, что постоянное или периодическое назначение ингибитора mTOR — рапамицина может увеличить продолжительность жизни зрелых мышей (Blagosklonny, 2011; Harrison et al., 2009). Примечательно, что рапамицин воздействует на несколько аспектов старения у мышей (Wilkinson et al., 2012). Увеличивающий продолжительность жизни эффект рапамицина строго зависит от индукции аутофагии у дрожжей, червей и мух (Bjedov et al., 2010; Rubinsztein et al., 2011). Однако похожих открытий в отношении эффектов рапамицина на старение млекопитающих еще не было сделано, а объяснить влияние рапамицина на продолжительность жизни можно другими механизмами (см «Нарушение распознавания питательных веществ»), такими как ингибирование рибосомальной S6 протеинкиназы 1 (S6K1), вовлеченной в синтез белка (Selman et al., 2009). Спермидин – другой индуктор макроаутофагии, который, в отличие от рапамицина, не имеет иммуносупрессивного побочного эффекта, также продлевает жизнь дрожжам, мухам и червям путем индукции аутофагии (Eisenberg et al., 2009). Добавление в пищу смеси полиаминов, содержащей спермидин, или обеспечение работы полиамин-продуцирующей флоры кишечника увеличивает продолжительность жизни у мышей (Matsumoto et al., 2011; Soda et al., 2009). Добавление в пищу ω-6 полиненасыщенных жирных кислот также увеличивает продолжительность жизни мух путем активации аутофагии (O’Rourke et al., 2013).
Что касается протеасом, активация EGF-сигналинга удлиняет продолжительность жизни нематод, увеличивая экспрессию различных компонентов убиквитин-протеасомной системы (Liu et al., 2011a). Подобным образом, усиление протеасомной активности ингибиторами деубиквитилазы или протеасомными активаторами ускоряет удаление токсичных белков в культуре человеческих клеток (Lee et al., 2010) и увеличивает репликативную продолжительность жизни у дрожжей (Kruegel et al., 2011). В дополнение к этому увеличенная экспрессия протеасомной субъединицы RPN-6 транскрипционным фактором DAF-16 семейства FOXO, повышает резистентность к протеотоксическому стрессу и продолжительность жизни у C. elegans (Vilchez et al., 2012).
В общих чертах
Имеются свидетельства, что старение ассоциировано с дефектами системы протеостаза, а ее экспериментальное нарушение ведет к развитию возраст-зависимых патологий. Также существуют впечатляющие примеры генетических манипуляций, которые улучшают протеостаз и замедляют старение у млекопитающих (Zhang и Cuervo, 2008).
Нарушение распознавания питательных веществ
Соматотропная ось млекопитающих включает гормон роста (ГР), который продуцируется передней долей гипофиза, и его вторичный медиатор – инсулиноподобный фактор роста 1 (ИФР-1), продуцируемый в ответ на ГР клетками многих типов, чаще всего гепатоцитами. Внутриклеточный сигнальный путь ИФР-1 совпадает с таковым у инсулина, который сообщает клеткам о наличии глюкозы. По этой причине ИФР-1 и инсулиновый сигналинг известны как молекулярный путь «ИФР-1 и инсулинового сигналинга» (ИИС). Примечательно, что путь ИИС является самым эволюционно консервативным метаболическим путем, контролирующим старение, среди его многочисленных мишеней есть транскрипционные факторы семейства FOXO и белки комплекса mTOR, которые также вовлечены в старение и сохранены эволюцией (Barzilai et al., 2012; Fontana et al., 2010; Kenyon, 2010). Генетические полиморфизмы и мутации, которые ослабляют функции ГР, рецептора ИФР-1, рецептора инсулина или их внутриклеточные эффекторы, такие как AKT, mTOR и FOXO, связаны с долголетием и у людей, и у модельных организмов, дополнительно иллюстрируя значительное влияние трофических и биоэнергетических путей на долголетие (Barzilai et al., 2012; Fontana et al., 2010; Kenyon, 2010) (Рисунок 4А).
Подтверждая уместность нарушения распознавания питательных веществ как ключевого признака старения, ограничение в диете (ОД) увеличивает продолжительность жизни и здоровую продолжительность жизни у всех изученных эукариотических организмов, включая низших приматов (Colman et al., 2009; Fontana er al., 2010; Mattison et al., 2012).
Сигнальные пути инсулина и ИФР-1
Многочисленные генетические манипуляции, которые нарушают сигналинг на разных уровнях молекулярного пути ИИС, увеличивают продолжительность жизни червей, мух и мышей (Fontana et al., 2010). Генетический анализ показал, что этот метаболический путь опосредует часть положительных эффектов ОД на долголетие у червей и мух (Fontana et al., 2010). Среди нижележащих эффекторов в сигнальном пути ИИС наибольшее влияние на долголетие червей и мух оказывает транскрипционный фактор FOXO (Kenyon et al., 1993; Slack et al., 2011). У мышей есть 4 представителя семейства FOXO, однако эффекты их сверхэкспрессии на долголетие и их роль в регуляции увеличения здоровой продолжительности жизни посредством уменьшения активности ИИС еще не установлены.
Мышиный FOXO1 требуется для проявления эффекта опухолевой супрессии ОД (Yamaza et al., 2010), однако неизвестно, вовлечен ли этот фактор в ОД-опосредованное увеличение продолжительности жизни. Как недавно было показано, у мыши с избыточной экспрессией опухолевого супрессора PTEN отмечается общее снижение активности сигнального пути ИИС и повышение количества энергии, ассоциированное с улучшением митохондриального окисления и повышенной активностью бурого жира (Garcia-Cao et al., 2012; Ortega-Molina et al., 2012). Линии других мышиных моделей со сниженной активностью ИИС, сверхэкспрессирующие Pten мыши, а также мыши с гипоморфным PI3K имеют более высокую продолжительность жизни (Foukas et al., 2013; Ortega-Molina et al., 2012).
Парадоксально, но уровни ГР и ИФР-1 снижаются как во время нормального старения, так и в мышиных моделях с преждевременным старением (Schumacher et al., 2008). Таким образом, снижение активности ИИС является общей характеристикой нормального и ускоренного старения, тогда как постоянно сниженная активность ИИС увеличивает продолжительность жизни. Эти с виду противоречащие друг другу наблюдения могут быть объяснены в рамках одной теории, по которой снижение активности ИИС отражает защитный ответ, нацеленный на минимизацию клеточного роста и метаболизма в контексте системного повреждения (Garinis et al., 2008). Согласно этой точке зрения, организмы с конституитивно сниженной активностью ИИС могут выживать дольше, потому как они имеют более низкую скорость клеточного роста и метаболизма и, как следствие, более низкий темп накопления клеточных повреждений. По той же самой причине физиологически или патологически постаревшие организмы стремятся снизить активность ИИС в попытке увеличить продолжительность жизни. Однако, защитные ответы, направленные против старения, сами имеют риск стать вредными и ускорить его, к этой концепции мы будем возвращаться в следующих разделах. Так, крайне низкие уровни ИИС несовместимы с жизнью, чему служат примером нулевые мутации PI3K или AKT киназ, которые летальны еще на эмбриональной стадии (Renner и Camero, 2009). Также у прогероидных мышей с очень низким уровнем ИФР-1 были случаи, когда дополнительный прием ИФР-1 мог замедлять преждевременное старение (Marino et al., 2010).
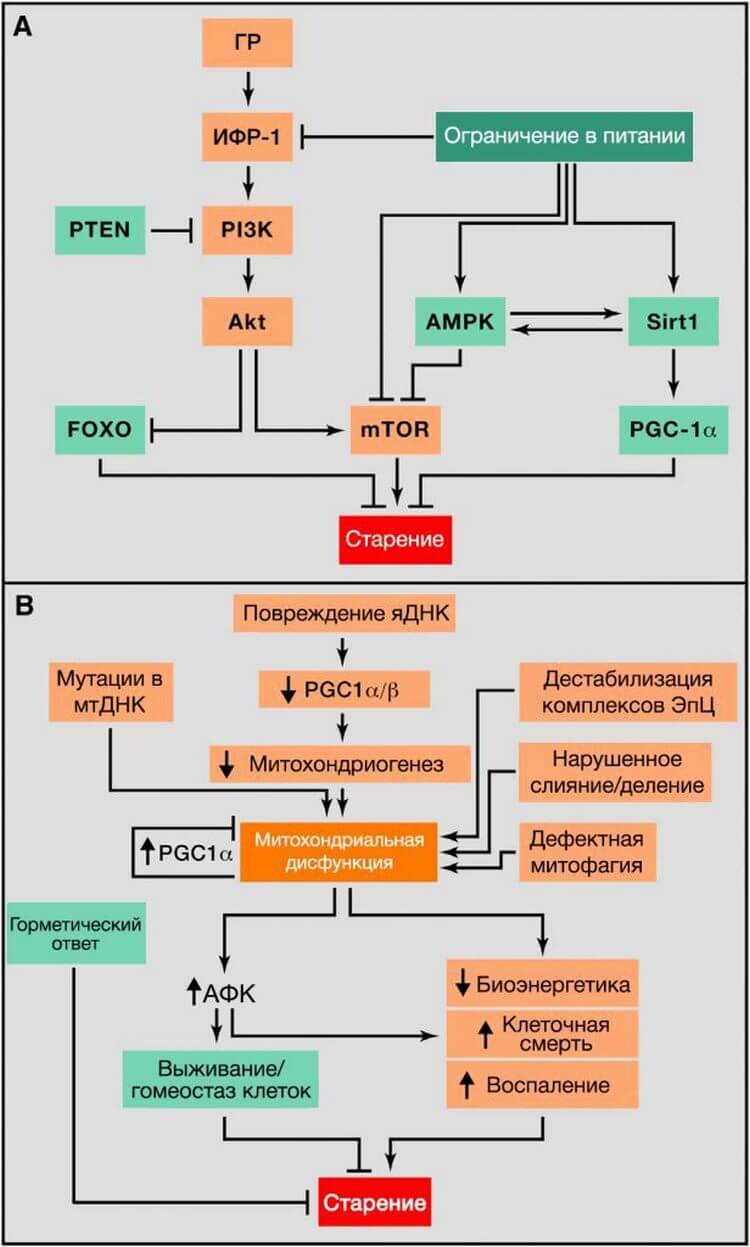
Рисунок 4. Метаболические альтерации
(A) Нарушение распознавания питательных веществ. Обзор соматотропной оси, включающей сигнальные пути гормона роста (ГР) и инсулиноподобного фактора роста 1 (ИФР-1), и ее связь между ограничением в питании и старением. Способствующие старению молекулы выделены оранжевым цветом, замедляющие старение молекулы выделены зеленым цветом.
(B) Митохондриальная дисфункция. Функция митохондрий нарушается в связи с возраст-ассоциированным накоплением мутаций в мтДНК, ослабляющих митохондриогенез, дестабилизирющих работу цепи переноса электронов (ЭТЦ), приводящих к изменению в митохондриальной динамике или ошибочной оценке качества митофагии. Вызванный стрессом сигнал и митохондриальная дисфункция приводят к увеличению АФК, что, при их относительно невысокой концентрации, приводит к активации сигналов выживания и восстановлению клеточного гомеостаза, но при продолжительном и/или высоком уровне АФК, приводит к старению. Точно так же, легкое митохондриальное повреждение может вызвать горметическую реакцию (митогормезис), которая запускает адаптивные компенсаторные ответы.
Другие системы распознавания питательных веществ: mTOR, AMPK и сиртуины
В дополнение к молекулярному пути ИИС, который участвует в контроле метаболизма глюкозы, в фокусе текущих исследований находятся еще три взаимосвязанные системы распознавания питательных веществ: mTOR, которая распознает высокие концентрации аминокислот; AMPK, которая реагирует на низкоэнергетические состояния, выявляя высокие уровни AMP; и сиртуины, которые реагируют на низкоэнергетические состояния, распознавая высокие уровни НАД+ (Houtkooper et al., 2010) (Рисунок 4А).
mTOR-киназа является частью двух мультипротеиновых комплексов: mTORC1 и mTORC2, которые регулируют практически все аспекты анаболизма (Laplante и Sabatini, 2012). Генетическая деактивация mTORC1 увеличивает продолжительность жизни и ослабляет положительные эффекты ОД у дрожжей, червей и мух, что свидетельствует о том, что ингибирование mTOR фенокопирует ОД (Johnson et al., 2013). У мышей применение рапамицина увеличивает продолжительность жизни, он считается самым надежным химическим средством для продления жизни у млекопитающих (Harrison et al., 2009). У генетически модифицированных мышей с низкой активностью mTORC1, но нормальной активностью mTORC2 отмечается увеличенная продолжительность жизни (Lamming et al., 2012), кроме того, мыши, дефицитные по S6K1 (основной субстрат mTORC1), также являются долгожителями (Selman et al., 2009). Таким образом, деактивация mTORC1/S6K1 является главным медиатором долголетия млекопитающих в отношении к mTOR. Более того, с возрастом увеличивается активность mTOR в нейронах гипоталамуса мыши, что вносит вклад в возраст-зависимое ожирение, которое можно вылечить прямой инфузией рапамицина в гипоталамус (Yang et al., 2012). Эти наблюдения, включая сигнальный путь ИИС, указывают на то, что избыточная трофическая и анаболическая активности, опосредуемые этими метаболическими путями, являются главными факторами, ускоряющими старение. И хотя ингибирование активности TOR имеет очевидные благоприятные эффекты на процесс старения, оно также имеет и такие нежелательные побочные эффекты, как нарушение заживления ран, инсулинорезистентность, катаракта и тестикулярная дегенерация у мышей (Wilkinson et al., 2012). Поэтому для того, чтобы понять на каком уровне благоприятные и повреждающие эффекты ингибирования TOR все еще могут быть отделены друг от друга, будет важным определить вовлеченные в этот процесс механизмы.
Другие две системы распознавания нутриентов – AMPK и сиртуины – по отношению к ИИС и mTOR действуют в обратном направлении: они сигнализируют о недостатке нутриентов и катаболизме, а не о наличии нутриентов и анаболизме. Соответственно, их повышенная экспрессия способствует здоровому старению. Активация AMPK имеет множество эффектов на метаболизм и, что примечательно, отключает mTORC1 (Alers et al., 2012). Есть доказательства, указывающие на то, что активация AMPK после применения метформина может приводить к увеличению продолжительности жизни у червей и мышей (Anisimov et al., 2011; Mair et al., 2011; Onken and Driscoll, 2010). Роль сиртуинов в регуляции продолжительности жизни была оговорена выше (см. “Эпигенетические альтерации). Кроме того, SIRT1 может деацетилировать и активировать PPARg коактиватор 1a (PGC-1a) (Rodgers et al., 2005). PGC-1a регулирует сложный метаболический ответ, который включает митохондриогенез, усиление антиоксидантной защиты и усиленное окисление жирных кислот (Fernandez-Marcos and Auwerx, 2011). Кроме того, SIRT1 и AMPK могут быть включены в цепь положительной обратной связи, объединяя оба сенсора низкоэнергетических состояний в единую систему (Price et al., 2012).
В общих чертах
Все имеющиеся на данный момент свидетельства подтверждают, что анаболический сигналинг ускоряет старение, а катаболический сигналинг увеличивает продолжительность жизни (Fontana et al., 2010). Кроме того, такие фармакологические манипуляции, которые имитируют состояние ограниченной доступности нутриентов, как применение рапамицина, могут увеличивать продолжительность жизни у мышей (Harrison et al., 2009).
Митохондриальная дисфункция
При старении клеток и организмов эффективность дыхательной цепи обычно снижается, и, как следствие, увеличивается утечка электронов и снижается генерация АТФ (Green et al., 2011) (Рисунок 4B). Долгое время считалось, что существует связь между митохондриальной дисфункцией и старением, но определение деталей такой связи остается основной задачей исследования старения.
Активные формы кислорода
Митохондриальная свободнорадикальная теория старения предполагает, что при старении наблюдается прогрессивная дисфункция митохондрий, которая приводит к повышенной выработке АФК, которые в свою очередь вызывают дальнейшее ухудшение работы митохондрий и глобальные повреждения клеток (Harman, 1965). Многочисленные данные подтверждают роль АФК в старении, но мы сфокусируемся на исследованиях последних 5 лет, благодаря которым произошла значительная переоценка митохондриальной свободнорадикальной теории старения (Hekimi et al., 2011). Особенно важным стало неожиданное наблюдение, что повышение АФК может увеличивать продолжительность жизни у дрожжей и C. elegans (Doonan et al., 2008; Mesquita et al., 2010; Van Raamsdonk and Hekimi, 2009). Также было замечено, что генетические манипуляции у мышей, которые приводят к увеличению АФК в митохондриях и оксидативным повреждениям, не ускоряют старение (Van Remmen et al., 2003; Zhang et al., 2009), а мыши с усиленной антиоксидантной защитой не живут дольше (Pe´rez et al., 2009), и, наконец, генетические манипуляции, которые нарушают функции митохондрий, не вызывают увеличения АФК-зависимого старения. (Edgar et al., 2009; Hiona et al., 2010; Kujoth et al., 2005; Trifunovic et al., 2004; Vermulst et al., 2008). Эти и другие данные подготовили почву для переосмысления роли АФК в процессах старения (Ristow and Schmeisser, 2011). Действительно, в сфере внутриклеточного сигналинга, развивающейся параллельно и автономно от работ по изучению повреждающих эффектов АФК, накоплено много свидетельств в пользу роли АФК в запуске пролиферации и выживания в ответ на физиологические сигналы и условия стресса (Sena and Chandel, 2012). Две линии доказательств могут быть согласованы, если рассматривать АФК как вызванный стрессом сигнал к выживанию, концептуально схожий с АМФ или НАД+ (см. «Нарушение распознавания питательных веществ»). В этом отношении первичным эффектом АФК будет активация компенсаторных гомеостатических ответов. С увеличением хронологического возраста, увеличивается клеточный стресс и повреждения, и параллельно, в попытке выжить, увеличивается уровень АФК. После достижения определенного порога АФК перестают выполнять свои гомеостатические функции и, наоборот, усугубляют, а не смягчают, возраст-ассоциированные повреждения (Hekimi et al., 2011). Эта новая концептуальная модель может помочь объединить, казалось бы, противоречивые данные о позитивных, негативных и нейтральных эффектах АФК на старение.
Митохондриальная целостность и биогенез
Дисфункциональные митохондрии могут вносить свой вклад в старение независимо от АФК, что подтверждается экспериментами с мышами, дефицитными по гамма-ДНК-полимеразе (Edgar et al., 2009; Hiona et al., 2010) (см. «Нестабильность генома»). В этом могут быть задействованы многие механизмы; например, нарушение работы митохондрий может затронуть апоптотический сигналинг за счет повышения склонности митохондрий к пермеабилизации в ответ на стресс (Kroemer et al., 2007) и запуска воспалительных реакций, обеспечивая АФК-опосредованную и/или облегченную пермеабилизацией активацию инфламмосом (Green et al., 2011). Также митохондриальная дисфункция может напрямую влиять на клеточный сигналинг и взаимодействие между органеллами, затрагивая область взаимодействия внешней митохондриальной мембраны и эндоплазматического ретикулума (Raffaello and Rizzuto, 2011).
Сниженная с возрастом эффективность митохондриальной биоэнергетики может быть результатом работы многих сходных механизмов, включая сниженный биогенез митохондрий, например, вследствие укорочения теломер у теломеразо-дефицитных мышей, с последующей р53 опосредованной репрессией PGC-1a и PGC-1b (Sahin and DePinho, 2012). Ухудшение работы митохондрий также происходит при физиологическом старении у мышей дикого типа и может быть частично восстановлено активацией теломеразы (Bernardes de Jesus et al., 2012). SIRT1 регулирует митохондриальный биогенез через процесс, включающий в себя транскрипционный коактиватор PGC-1a (Rodgers et al., 2005) и удаление поврежденных митохондрий в процессе аутофагии (Lee et al., 2008). SIRT3, который является основной митохондриальной деацетилазой (Lombard et al., 2007), направлен на многие ферменты, вовлеченные в энергетический метаболизм, в том числе и на компоненты дыхательной цепи, цикл трикарбоновых кислот, кетогенез и бета-окисление жирных кислот (Giralt and Villarroya, 2012). SIRT3 может также напрямую контролировать продукцию АФК, деацетилируя марганцевую супероксиддисмутазу, основной митохондриальный антиоксидантный фермент (Qiu et al., 2010; Tao et al., 2010). В совокупности эти наблюдения поддерживают идею, что теломеры и сиртуины могут контролировать митохондриальную функцию и таким образом играть защитную роль против возраст-зависимых заболеваний.
Другие причинные механизмы дефектной биоэнергетики включают накопление мутаций и делеций в мтДНК, окисление митохондриальных белков, дестабилизацию макромолекулярной организации (супер)комплексов дыхательной цепи, изменения в липидном составе митохондриальных мембран, изменения в митохондриальной динамике, происходящие из-за нарушения баланса между делением и слиянием и плохим контролем качества митофагии – органелло-специфичной формы макроаутофагии, которая захватывает дисфункциональные митохондрии для протеолитической деградации (Wang and Klionsky, 2011). Комбинация увеличения количества повреждений и снижения обновления митохондрий в результате сниженного биогенеза и нарушений удаления продуктов распада может вносить свой вклад в старение (Рисунок 4В).
Интересно, что тренировки на выносливость и периодические ограничения в питании могут увеличивать продолжительность здоровой жизни, защищая митохондрии от дегенерации (Castello et al., 2011; Safdar et al., 2011). Хочется думать, что эти положительные эффекты, по крайней мере, частично опосредованы индукцией аутофагии, так как и тренировки на выносливость, и периодические ограничения в питании служат мощными ее триггерами (Rubinsztein et al., 2011). Однако индукция аутофагии является не единственным механизмом, посредством которого здоровый образ жизни может замедлять старение, так как при соблюдении определённого режима ограничения питания могут быть активированы дополнительные молекулярные пути, ведущие к долголетию (Kenyon, 2010).
Митогормезис
Митохондриальная дисфункция при старении также связана с гормезисом — концепцией, на которой сошлись несколько направлений исследований (Calabrese et al., 2011). Согласно этой концепции применение слаботоксичных препаратов запускает благоприятные компенсаторные ответы, которые не только восстанавливают пусковые повреждения, но и делают клетку более устойчивой к повреждениям. Таким образом, хотя серьезная дисфункция митохондрий ведет к патологии, небольшой респираторный дефицит может увеличивать продолжительность жизни, возможно, благодаря горметическому эффекту (Haigis и Yankner, 2010). Горметические реакции могут запускать митохондриальный защитный ответ как в самой ткани с дефектными митохондриями, так и в удаленных тканях, как было показано у C. elegans (Durieux et al., 2011). Есть убедительные доказательства, что такие соединения, как метформин и ресвератрол, являются слабыми митохондриальными ядами, которые вызывают низкоэнергетическое состояние, характеризующееся увеличением количества АМФ и активацией AMPK (Hawley et al., 2010). Немаловажно, что метформин увеличивает продолжительность жизни C. elegans путем индукции компенсаторного стресс-ответа, опосредованного AMPK и главным регулятором антиоксидантной системы NRF2 (Onken и Driscoll, 2010). Последние исследования также показали, что метформин замедляет старение у червей, изменяя у их кишечного микробиома метаболизм фолата и метионина (Cabreiro et al., 2013). Касательно млекопитающих, метформин может увеличивать продолжительность жизни мышей, если назначается с первых месяцев жизни (Anisimov et al., 2011). В отношении ресвератрола и активатора сиртуинов SRT1720 имеются убедительные доказательства, что через PGC-1a зависимый механизм они защищают от метаболического повреждения и улучшают дыхание митохондрий (Baur et al., 2006; Feige et al., 2008; Lagouge et al., 2006; Minor et al., 2011), хотя при условиях нормального питания ресвератрол не способен увеличивать продолжительность жизни мышей (Pearson et al., 2008; Strong et al., 2013). О влиянии PGC-1a на долголетие говорит также наблюдение, что его сверхэкспрессия, ассоциированная с улучшенной активностью митохондрий, увеличивает продолжительность жизни у Drosophila (Rera et al., 2011). Наконец, разобщение процессов окислительного фосфорилирования в митохондриях как генетически, через сверхэкспрессию белка UCP1, так и при назначении химического разобщителя 2,4-динитрофенола, может увеличивать продолжительность жизни у мух и мышей (Caldeira da Silva et al., 2008; Fridell et al., 2009; Gates et al., 2007; Mookerjee et al., 2010).
В общих чертах
Функционирование митохондрий оказывает значительное влияние на процессы старения. Митохондриальная дисфункция у млекопитающих может их ускорять (Kujoth et al., 2015; Trifunovic et al., 2004; Vermulst et al., 2008), однако остается неясным, обеспечивает ли повышение митохондриальной функции у млекопитающих, как, например, при митогормезисе, увеличение продолжительности жизни, хотя по имеющимся данным есть основания это предполагать.
Клеточное старение
Клеточная сенесценция может быть определена как прекращение клеточного деления с характерными изменениями фенотипа (Campisi и d’Adda di Fagagna, 2007; Collado et al., 2007; Kuilman et al., 2010) (Рисунок 5А). Этот феномен первоначально описан Хейфликом у последовательно пересеваемых культур человеческих фибробластов (Hayflick и Moorhead, 1961). Сегодня мы знаем, что старение, которое наблюдал Хейфлик, было вызвано укорочением теломер (Bodnar et al., 1998), однако есть и другие возраст-ассоциированные стимулы, независимо от них запускающие старение. В первую очередь это повреждение нетеломерных участков ДНК и депрессия локуса INK4/ARF, которые прогрессируют при хронологическом старении, а также способны вызывать сенесценцию клеток (Collado et al., 2007). Накопление сенесцентных клеток в стареющих тканях оценивается по косвенным маркерам, таким как повреждение ДНК. В некоторых исследованиях для обнаружения в тканях сенесцентных клеток определяют ассоциированную со старением бета-галактозидазу (САБГ) (Dimri et al., 1995). Следует отметить, что при детальном параллельном определении в печени мышей количества САБГ и повреждений ДНК общее количество сенесцентных клеток составило ~8% у молодой мыши и ~17% у очень старой (Wang et al., 2009). Аналогичные результаты были получены для кожи, легких и селезенки, однако в сердце, скелетных мышцах и почках никаких изменений не наблюдалось (Wang et al., 2009). Основываясь на этих данных, можно утверждать, что сенесценция свойственна не всем тканям стареющих организмов. В случае сенесцентных опухолевых клеток есть бесспорные свидетельства того, что они находятся под строгим иммунным надзором и эффективно удаляются путём фагоцитоза (Hoenicke and Zender, 2012; Kang et al., 2011; Xue et al., 2007). Предположительно, накопление сенесцентных клеток при старении может характеризоваться увеличением скорости генерации сенесцентных клеток и/или снижением скорости их удаления, например, вследствие ослабленного иммунного ответа.
Так как при старении число сенесцентных клеток растет, предполагается, что сенесцентность вносит в старение определённый вклад. Однако эта точка зрения недооценивает предполагаемую главную роль сенесцентности – предотвращение распространения поврежденных клеток и запуск их уничтожения иммунной системой. Таким образом, существует вероятность, что сенесцентность является важным компенсаторным механизмом, который избавляет ткани от поврежденных и потенциально онкогенных клеток. Данная контрольная точка клетки, однако, требует эффективной системы замены для восстановления числа клеток, включающей удаление сенесцентных клеток и мобилизацию клеток-предшественниц. При старении организма этот круговорот может стать неэффективным, или может истощаться регенеративная способность клеток-предшественниц, что в итоге приводит к усилению повреждения, способствующего старению (Изображение 5А).
В последние годы все больше обращают внимание на то, что сенесцентные клетки претерпевают значительные альтерации своего секретома, в котором становится особенно много провоспалительных цитокинов и матричных металлопротеиназ и который часто называют «ассоциированным со старением секреторным фенотипом» (Kuilman et al., 2010; Rodier and Campisi, 2011). Этот провоспалительный секретом может вносить свой вклад в старение (см. «Межклеточная коммуникация»).
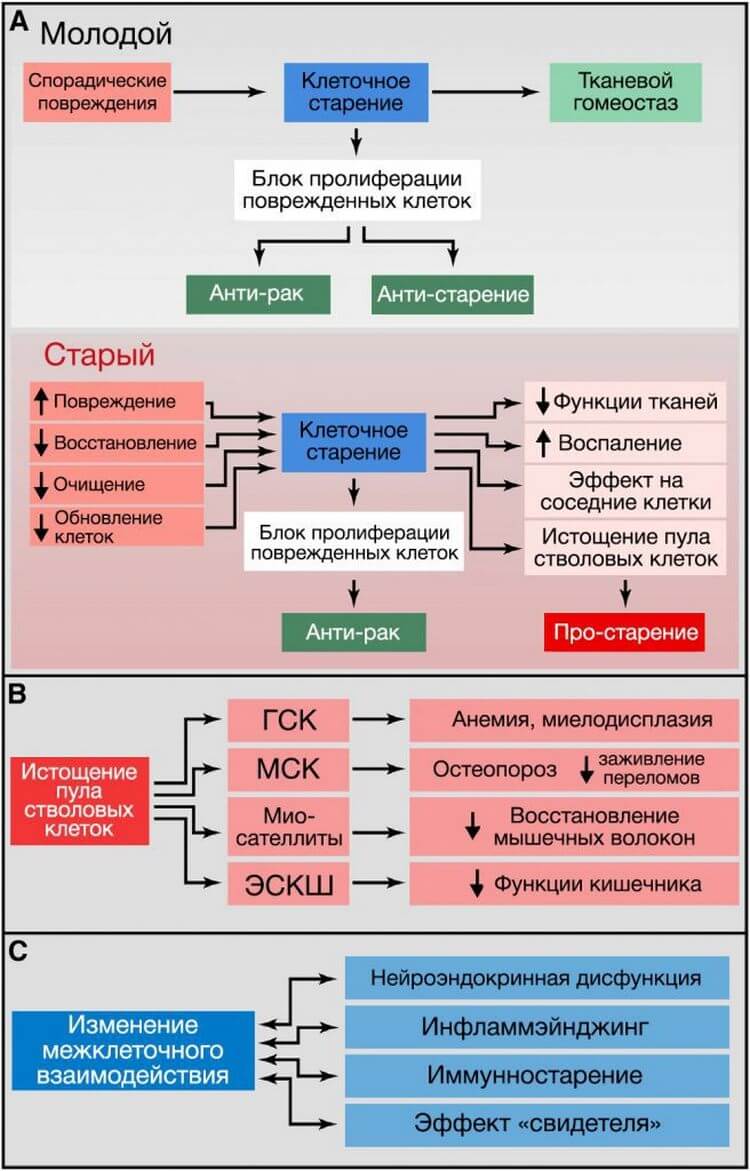
Рисунок 5. Клеточная сенесценция, истощение пула стволовых клеток и изменение межклеточного взаимодействия
(A) Клеточная сенесценция. У молодых организмов клеточное старение предотвращает пролиферацию поврежденных клеток, таким образом предотвращая развитие рака и способствуя нормальному гомеостазу ткани. В старых организмах распространяющееся повреждение и неполноценный клиренс стареющих клеток ведет к их накоплению, что приводит ко множеству неблагоприятных эффектов, способствующих старению.(B) Истощение стволовых клеток. На изображении показаны последствия истощения гематопоэтических стволовых клеток (ГСК), мезенхимальных стволовых клеток (МСК), миосателлитоцитов и эпителиальных стволовых клеток кишечника (ЭСКШ).
(C) Изменение межклеточного взаимодействия. Связанные со старением примеры изменения межклеточного взаимодействия.
INK4a/ARF локус и p53
Помимо повреждения ДНК, одним из сопряжённых со старением факторов стресса является избыточный митогенный сигналинг. В недавнем отчёте перечислено 50 онкогенных и митогенных альтераций, способных вызывать сенесцентность (Gorgoulis and Halazonetis, 2010). Выросло также число механизмов, запускающих сенесцентность в ответ на многочисленные онкогенные повреждения, однако изначально обнаруженные p16INK4a/Rb и p19ARF/p53 метаболические пути остаются самыми важными (Serrano et al., 1997). Релевантность этих молекулярных путей при старении становится еще более значимой, если принять во внимание, что почти во всех проанализированных тканях человека и мыши уровень p16INK4a (и в меньшей степени p19ARF) коррелирует с хронологическим возрастом (Krishnamurthy et al., 2004; Ressler et al., 2006). Это единственный известный ген, чья экспрессия среди такого разнообразия тканей и видов так сильно коррелирует с хронологическим старением, и эта экспрессия в старой ткани может быть, в среднем, до 10 раз выше, чем в молодой. И p16INK4a, и p19ARF кодируются одним и тем же генетическим локусом INK4a/ARF. Последний мета-анализ более чем 300 исследований полногеномных ассоциаций (ИПГА) показал, что INK4a/ARF локус генетически связан с наибольшим количеством возраст-ассоциированных патологий, включая несколько типов сердечно-сосудистых заболеваний, диабет, глаукому и болезнь Альцгеймера (Jeck et al., 2012). Эти наблюдения позволяют считать INK4a/ARF локус наиболее изученным генетическим фактором, контролирующим старение человека и развитие возраст-зависимых патологий. Остается нерешенным важный вопрос, кодируют ли ассоциированные с болезнью аллели INK4a/ARF приобретение или потерю функции.
Критическая роль p16INK4a и p53 в индуцировании клеточной сенесцентности дает основания предполагать, что вызванная p16INK4a или p53 сенесцентность вносит вклад в физиологическое старение. Согласно этой точке зрения, вызывающая старение активность p16INK4a и p53 является разумной платой за их участие в подавлении роста опухолей. Это подтверждается тем, что мутантные мыши с преждевременным старением из-за избыточного и непрерывного повреждения имеют высокий уровень сенесцентности, однако их прогероидный фенотип улучшается при удалении p16INK4a или p53. То же происходит у мышей, дефицитных по BRCA1 (Cao et al., 2003), модельных мышей с ПСХГ (Varela et al., 2005) и мышей с дефектом стабильности хромосом из-за гипоморфной мутации BubR1 (Baker et al., 2011). Однако другие данные предполагают более сложную модель. В противоположность предполагаемой роли этих генов в старении, у мышей с незначительным системным увеличением экспрессии опухолевых супрессоров p16INK4a, p19ARF или p53 отмечается увеличенная продолжительность жизни, не связанная с более низкой вероятностью развития рака (Matheu et al., 2007, 2009). Также удаление p53 ухудшает фенотип некоторых прогероидных мутантных мышей (Begus-Nahrmann et al., 2009; Murga et al., 2009; Ruzankina et al., 2009). Как и в случае, рассмотренном выше сенесцентности, активация p53 и INK4a/ARF может рассматриваться как благоприятный компенсаторный ответ, нацеленный на предупреждение распространения поврежденных клеток и последствий этого распространения в виде старения и рака. Однако, когда повреждения слишком обширны, регенеративная способность тканей может быть истощена или подавлена, и в этих критических условиях ответы p53 и INK4a/ARF могут стать губительными и ускорить старение.
В общих чертах
Мы предполагаем, что клеточная сенесцентность является целесообразным компенсаторным ответом на повреждение, который становится вредоносным и ускоряет старение, когда ткани теряют свой регенеративный потенциал. Принимая во внимание эти аспекты, невозможно однозначно ответить на вопрос, удовлетворяет ли клеточная сенесцентность третьему критерию ключевого признака. Умеренная стимуляция вызывающих сенесцентность метаболических путей опухолевой супрессии может увеличивать продолжительность жизни (Matheu et al., 2007, 2009), и в то же время удаление сенесцентных клеток в экспериментальной модели прогерии замедляет развитие возраст-зависимых патологий (Baker et al., 2011). Таким образом, оба вмешательства, которые являются принципиально противоположными, способны увеличивать продолжительность здоровой жизни.
Клеточное старение
Снижение регенеративного потенциала тканей является одной из наиболее очевидных характеристик старения (Изображение 5В). Например, с возрастом снижается гемопоэз, что приводит к снижению продукции адаптивных иммунных клеток – процессу, называемому иммуностарением – и к повышенному риску развития анемии и миелоидных злокачественных образований (Shaw et al., 2010). Подобная функциональная потеря стволовых клеток обнаружена практически во всех компартментах взрослых стволовых клеток, включая передний мозг мыши (Molofsky et al., 2006), кость (Gruber et al., 2006) и мышечные ткани (Conboy and Rando, 2012). Исследования, проведенные на старых мышах, позволили обнаружить снижение активности клеточного цикла гемопоэтических стволовых клеток (ГСК) – старые ГСК претерпевали меньшее количество клеточных делений, чем молодые ГСК (Rossi et al., 2007). Снижение коррелирует с накоплением повреждений ДНК (Rossi et al., 2007)и с гиперэкспрессией белков-ингибиторов клеточного цикла, таких как p16INK4a (Janzen et al., 2006). Фактически, старые INK4a-/- ГСК лучше приживаются и имеют более высокую активность клеточного цикла, по сравнению со старыми ГСК дикого типа (Janzen et al., 2006). Укорочение теломер при старении также является важной причиной угасания функций стволовых клеток и уменьшения их количества во многих тканях (Flores et al., 2005; Sharpless and DePinho, 2007). Всё это лишь отдельные примеры из огромной системы реакций, в которой ухудшение работы и уменьшение количества стволовых клеток является следствием интеграции множества различных повреждений.
Хотя снижение пролиферации стволовых и прогениторных клеток, очевидно, вредит долговременному поддержанию работы организма, избыточная их пролиферация также может быть вредной за счет ускорения истощения их клеточной ниши. Важность покоя стволовых клеток для долговременного сохранения их функциональности была убедительно продемонстрирована на примере стволовых интестинальных клеток Drosophila, избыточная пролиферация которых ведет к истощению и преждевременному старению (Rera et al., 2011). Похожая ситуация встречается и у p21-нулевых мышей, у которых наблюдается преждевременное истощение ГСК и нейральных стволовых клеток (Cheng et al., 2000; Kippin et al., 2005). В этом отношении, индукция INK4a при старении (см. «Клеточная сенесценция») и снижение ИФР-1 в сыворотке (см. «Нарушение распознавания питательных веществ») могут отражать попытку организма сохранить покой стволовых клеток. Кроме того, недавние исследования показали, что увеличение активности сигналинга FGF2 в нише старых мышечных стволовых клеток приводит к выходу из состояния покоя и, в конечном итоге, к истощению стволовых клеток и сниженной способности к регенерации, тогда как подавление данного сигналинга предотвращает этот дефект (Chakkalakal et al., 2012). Это открывает перспективы для создания стратегий, нацеленных на ингибирование сигналинга FGF2, которое позволит уменьшить истощение стволовых клеток при старении. По тому же принципу ОП улучшает функцию интестинальных и мышечных стволовых клеток (Cerletti et al., 2012; Yilmaz et al., 2012).
Важным вопросом является определение роли внутриклеточных и внеклеточных молекулярных путей в снижении функции стволовых клеток (Conboy and Rando, 2012). Новые работы свидетельствуют в пользу последних. В частности, трансплантация стволовых клеток, полученных из мышцы молодых мышей, прогероидным мышам увеличивает продолжительность жизни и уменьшает дегенеративные изменения у этих животных даже в тканях, в которых донорские клетки не обнаруживаются, что дает основание предполагать, что их терапевтический эффект может происходить благодаря системному действию секретируемых факторов (Lavasani et al., 2012). Более того, эксперименты с парабиозом показали, что сниженные функции нейральных и мышечных стволовых клеток могут быть восстановлены системными факторами, полученными у молодых мышей (Conboy et al., 2005; Villeda et al., 2011).
Также исследуют фармакологические способы улучшения функционирования стволовых клеток. Так, ингибирование mTORC1 рапамицином может замедлять старение, улучшая протеостаз (см. «Нарушение протеостаза»), и, влияя на распознавание веществ (см. «Нарушенное распознавание питательных веществ»), может улучшать работу стволовых клеток эпидермиса, гемопоэтической системы и кишечника (Castilho et al., 2009; Chen et al., 2009; Yilmaz et al., 2012). Это иллюстрирует сложность в понимании основы механизмов активности рапамицина против старения и подчеркивает взаимосвязанность обсуждаемых здесь различных ключевых признаков старения. Также стоит отметить возможность омоложения человеческих сенесцентных клеток фармакологическим ингибированием ГТФазы CDC42, чья активность увеличивается при старении ГСК (Florian et al., 2012).
В общих чертах
Истощение стволовых клеток является следствием множества различных возраст-ассоциированных повреждений, и, вероятно, является одной из основных причин старения тканей и организма. Последние многообещающие исследования дают основания предполагать, что омоложение стволовых клеток может обратимо воздействовать на фенотип старения на организменном уровне (Rando and Chang, 2012).
Измененная межклеточная коммуникация
Помимо автономных изменений в клетке, старение также включает изменения на уровне межклеточной коммуникации: эндокринной, нейроэндокринной или нервной (Laplante and Sabatini, 2012; Rando and Chang, 2012; Russell and Kahn, 2007; Zhang et al., 2013) (Изображение 5С). Таким образом, регуляция нейрогормонального сигналинга (например, ренин-ангиотензинового, адренергического, инсулин-ИФР1 сигналинга) при старении склонна нарушаться вследствие того, что усиливаются воспалительные реакции, снижается надзор иммунитета за патогенами и предопухолевыми клетками, и меняется состав около- и внеклеточной среды.
Воспаление
Важным возраст-ассоциированным изменением в межклеточной коммуникации является «инфламмэйнджинг», то есть, вялотекущий провоспалительный фенотип, сопутствующий старению у млекопитающих (Salminen et al., 2012). Инфламмэйджинг может быть результатом многих причин, таких как накопление провоспалительных повреждений ткани, неспособность всё слабеющей иммунной системы эффективно удалять патогены и дисфункциональные клетки организма, склонность сенесцентных клеток секретировать провоспалительные цитокины (см. «Клеточная сенесценция»), повышенная активация транскрипционного фактора NF-kB или нарушения в процессе аутофагии (Salminen et al., 2012). Эти изменения приводят к повышенной активации инфламмосомы NLRP3 и других провоспалительных путей, что в итоге ведет к повышенной продукции IL-1b, фактора некроза опухоли и интерферонов (Green et al., 2011; Salminen et al., 2012). Воспаление также вовлечено в патогенез ожирения и диабета 2 типа – двух состояний, которые в человеческой популяции коррелируют со старением, а также усугубляют его (Barzilai et al., 2012). Похожим образом, патологический воспалительный ответ играет критическую роль в развитии атеросклероза (Tabas, 2010). Недавнее открытие, что возраст-зависимое воспаление подавляет функции эпидермальных стволовых клеток (Doles et al., 2012), подтверждает сложную взаимосвязь между различными ключевыми признаками, усиливающими процесс старения. Параллельно инфламмэйджингу снижается функция адаптивной иммунной системы (Deeks, 2011). Это иммуностарение может на системном уровне усугублять фенотип старения из-за неспособности иммунной системы удалять инфекционные агенты, зараженные клетки и клетки на грани опухолевой трансформации. Более того, одной из функций иммунной системы является распознавание и удаление сенесцентных клеток (см. «Истощение стволовых клеток»), а также гиперплоидных клеток, которые накапливаются в стареющих тканях и в предопухолевых состояниях (Davoli and de Lange, 2011; Senovilla et al., 2012).
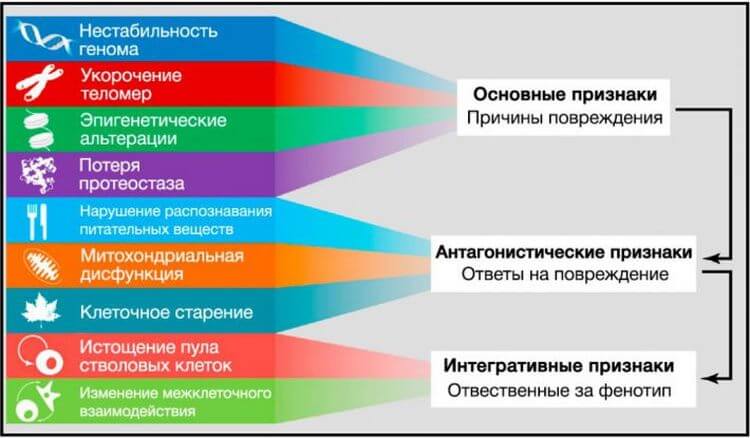
Рисунок 6. Функциональные взаимосвязи между признаками старения
Девять описанных признаков старения объединены в три категории.
(Верхушка) Признаки, которые, как полагают, являются основными причинами повреждения клеток.(Середина) Признаки, которые считают частью компенсаторных или антагонистических ответов на повреждение. Первоначально эти реакции направлены на уменьшение повреждения, но в итоге, при хроническом или интенсивном воздействии, приводят к повреждениям.(Основание) Интегративные признаки, которые являются конечным результатом признаков предыдущих двух групп и, в конечном счете, ответственны за снижение функций, связанное со старением.Глобальные исследования транскриптомного профиля стареющих тканей подчеркивают особую роль воспалительных метаболических путей в процессе старения (de Magalha˜ es et al., 2009; Lee et al., 2012). Избыточная активация метаболического пути NF-kB является одним из транскрипционных признаков старения, а умеренная экспрессия ингибитора NF-kB в стареющей коже трансгенных мышей вызывает омоложение фенотипа этой ткани, также как и восстановление транскрипционных признаков, характерных для молодого возраста (Adler et al., 2007). Подобным образом у мышей различных линий с ускоренным старением генетическое и фармакологическое ингибирование NF-kB сигналинга предотвращает развитие возраст-ассоциированных особенностей (Osorio et al., 2012; Tilstra et al., 2012). Новая связь между воспалением и старением возникла после недавнего открытия активации в ответ на воспаление и стресс NF-kB в гипоталамусе и запуска сигнального метаболического пути, приводящего к сниженной продукции нейронами гонадотропин-рилизинг-гормона (ГнРГ) (Zhang et al., 2013). Снижение ГнРГ может вносить вклад в многочисленные возраст-зависимые изменения, такие как хрупкость костей, слабость мышц, атрофия кожи и сниженный нейрогенез. Закономерно, применение ГнРГ предотвращает возрастное нарушение нейрогенеза и замедляет старение у мышей (Zhang et al., 2013). Эти наблюдения дают основание предполагать, что гипоталамус за счет ГнРГ-опосредованных нейроэндокринных эффектов может модулировать системное старение, интегрируя запущенные NF-kB воспалительные ответы.Ещё одно свидетельство связи между воспалением и старением in vivo найдено в ходе работы над фактором распада мРНК AUF1, участвующего в прекращении воспалительного ответа за счет опосредованной цитокинами деградации мРНК (Pont et al., 2012). AUF1-дефицитные мыши имеют выраженную клеточную сенесценцию и фенотип преждевременного старения, которые могут быть восстановлены экспрессией этого РНК-связывающего фактора. Интересно, что в дополнение к регуляции распада мРНК с участием воспалительных цитокинов, AUF1 играет роль в поддержании длины теломер за счет активации экспрессии каталитической субъединицы теломеразы TERT (Pont et al., 2012), снова демонстрируя, что один фактор может влиять на различные ключевые признаки старения.Аналогичная ситуация происходит с сиртуинами, которые также оказывают влияние на ассоциированный со старением воспалительный ответ. Несколько исследований показали, что путём деацетилирования гистонов и компонентов воспалительных сигнальных путей, таких как NF-kB, SIRT1 можно снизить экспрессию генов, связанных с воспалением (Xie et al., 2013). В соответствии с этими открытиями, снижение уровня SIRT1 коррелирует с развитием и прогрессированием многих воспалительных заболеваний, а фармакологическая активация SIRT1 у мышей может предотвратить развитие воспалительных ответов (Gillum et al., 2011; Yao et al., 2012; Zhang et al., 2010). SIRT2 и SIRT6 также могут препятствовать развитию воспалительного ответа путем деацетилирования субъединиц NF-kB и транскрипционной репрессии их генов-мишеней (Kawahara et al., 2009; Rothgiesser et al., 2010).
Другие типы межклеточной коммуникации
Накапливаются данные, подтверждающие, что возраст-зависимые изменения в одной ткани могут вести к возраст-специфичным нарушениям в других тканях, что объясняет межорганную координацию фенотипа старения. Наряду с воспалительными цитокинами имеются другие примеры «заразного старения» или эффекта «свидетеля», при котором сенесцентные клетки вызывают сенесценцию у соседних клеток через межклеточные щелевые контакты и межклеточные процессы, с участием АФК (Nelson et al., 2012). Микроокружение вносит свой вклад в развитие возраст-зависимых функциональных дефектов T-клеток CD4+, что было исследовано на мышиной модели с адаптивным переносом (Lefebvre et al., 2012). Манипуляции, увеличивающие продолжительность жизни и нацеленные только на одну ткань, могут замедлять процесс старения и в других тканях (Durieux et al., 2011; Lavasani et al., 2012; Toma´ s-Loba et al., 2008).
Восстановление дефектной межклеточной коммуникации
Есть несколько способов восстановления дефектной межклеточной коммуникации, также обусловливающей старение, которые включают генетические, пищевые и фармакологические мешательства, которые могут улучшить снижающуюся с возрастом эффективность межклеточных взаимодействий (Freije and Lo´ pez-Otı´n, 2012; Rando and Chang, 2012). Особый интерес представляют методики ОП для увеличения продолжительности жизни (Piper et al., 2011; Sanchez-Roman et al., 2012) и стратегии по омоложению, основанные на использовании системных факторов крови, определенных в экспериментах по парабиозу (Conboy et al., 2005; Loffredo et al., 2013; Villeda et al., 2011). Более того, краткосрочное назначение противовоспалительных препаратов, таких как аспирин, может увеличивать продолжительность жизни у мышей и способствовать здоровому старению у людей (Rothwell et al., 2011; Strong et al., 2008). Принимая во внимание то, что кишечный микробиом влияет на функционирование иммунной системы хозяина и обладает системными метаболическими эффектами, можно сделать вывод о возможности увеличения продолжительности жизни, манипулируя составом и функциональностью сложной и динамичной кишечной бактериальной экосистемы человеческого организма (Claesson et al., 2012; Ottaviani et al., 2011).
В общих чертах
Существуют веские доказательства того, что старение затрагивает не только клетки, но и влияет на общие изменения в межклеточной коммуникации, что дает возможность контролировать старение на этом уровне. Доказательством этого является омоложение с помощью системных факторов крови (Conboy et al., 2005; Loffredo et al., 2013; Villeda et al., 2011).
Заключения и перспективы
Все девять ключевых признаков старения, перечисленных в данном обзоре, можно разделить на три группы: основные признаки, антагонистические признаки и интегративные признаки (Рисунок 6). Общей характеристикой основных признаков является то, что они однозначно негативные. Это повреждение ДНК, включая хромосомальные анеуплоидии; мутации митохондриальной ДНК; укорочение теломер, эпигенетический сдвиг и дефектный протеостаз. В отличие от основных признаков, антагонистические вызывают разные эффекты в зависимости от интенсивности проявления. В небольшом количестве они оказывают благоприятное воздействие, но в большом они становятся вредными. К ним относится сенесценция, которая защищает организм от рака, но которая в избытке ускоряет старение. Подобным образом АФК опосредуют клеточный сигналинг и выживание, но при длительном превышении порога концентрации могут вызывать клеточные повреждения; также оптимальное распознавание питательных веществ и оптимальный анаболизм бесспорно необходимы для выживания, но их длительное пребывание в состоянии высокой активности может перерасти в патологию. Эти ключевые признаки могут рассматриваться в качестве защиты организма от повреждения или недостатка питательных веществ. Но когда они находятся в состоянии хронической активности, то больше не выполняют свою функцию, а провоцируют дальнейшее повреждение. Третья категория включает интегративные признаки – истощение стволовых клеток и нарушение межклеточной коммуникации – которые напрямую влияют на клеточный гомеостаз и функцию. Несмотря на взаимосвязанность между всеми ключевыми признаками, мы предлагаем определенную иерархию (Рисунок 6). Основные признаки могут быть триггерами, чьи негативные эффекты со временем накапливаются. Антагонистические признаки, являющиеся в принципе полезными, становятся вредными под влиянием основных признаков. Наконец, интегративные признаки проявляются, когда накопившиеся повреждения, вызванные основными и антагонистическими признаками, не могут быть компенсированы механизмами тканевого гомеостаза. Так как при старении ключевые признаки проявляются совместно и тесно связаны, то понимание причинно-следственной связи между ними является важным вопросом для дальнейших исследований.
Рисунок 7. Меры, которые могут способствовать продлению жизни человека
Изображены все девять признаков старения, а также возможные стратегии терапии, которые прошли экспериментальное подтверждение на мышах.
Определение ключевых признаков старения может помочь (1) построить основные принципы будущих исследований молекулярных механизмов старения и (2) создать терапевтические методики, которые позволят увеличить продолжительность здоровой жизни человека (Рисунок 7). Однако впереди еще много не отвеченных вопросов в отношении понимания этого комплексного биологического процесса (Martin, 2011; Miller, 2012). Быстрое развитие технологий секвенирования нового поколения может оказать большое влияние на исследование старения, облегчая изучение генетических и эпигенетических изменений, специфично накапливающихся в клетках стареющего организма (de Magalha˜ es et al., 2010; Gundry and Vijg, 2012). Эти технологии уже используются для определения полногеномного сиквенса долгоживущих индивидуумов, чтобы провести сравнительные геномные исследования между коротко- и долгоживущими линиями и видами животных, и чтобы проанализировать возраст-зависимые эпигенетические изменения в мельчайших деталях (Heyn et al., 2012; Kim et al., 2011; Sebastiani et al., 2011). Необходимы параллельные исследования in vivo с моделями мутантных животных с приобретением или потерей функции, которые позволят выйти за рамки коррелятивного анализа, и получить доказательства подкрепляющие роль ключевых факторов в процессе старения. Кроме того, чтобы понять причинно-следственную связь между процессами, которые сопровождают старение, и процессами, которые к нему ведут, требуется описание характера индивидуальных ключевых признаков с использованием подходов системной биологии (Gems and Partridge, 2013; Kirkwood, 2008). В дополнение к вышесказанному, молекулярный анализ взаимодействия генома и среды поможет идентифицировать мишени для лекарств, увеличивающих продолжительность жизни (de Magalha˜ es et al., 2012). Мы предполагаем, что более тщательно продуманные подходы помогут окончательно разрешить имеющиеся вопросы. Надеемся, что комбинирование этих подходов позволит детально понять механизмы, лежащие в основе ключевых признаков старения, и поможет разработать основные принципы терапии, направленной на увеличение продолжительности здоровой жизни и долголетия человека.
Понравилась статья? Поделитесь с друзьями на Facebook:


